SMA, identificate le alterazioni neurometaboliche che interferiscono con la comunicazione tra le cellule nervose.
I risultati di una ricerca italiana aprono la strada a nuovi approcci terapeutici per potenziare i farmaci oggi disponibili
Un network di ricercatori e clinici nel campo delle neuroscienze ha identificato gravi alterazioni biochimiche epatiche e neurometaboliche nella Atrofia Muscolare Spinale (SMA), una malattia genetica rara, che nella forma più grave provoca paralisi fino alla morte prematura dei bambini. I risultati dello studio, durato 4 anni e condotto su 33 pazienti pediatrici ed in modelli animali, sono stati pubblicati sulla rivista scientifica Communication Biology (Nature Group). La ricerca è stata sviluppata grazie ad una stretta collaborazione tra il CEINGE Biotecnologie Avanzate Franco Salvatore di Napoli, l’Università della Campania Luigi Vanvitelli, l’Università di Napoli Federico II, l’Università di Salerno, l’Università di Cagliari e l’Ospedale Bambino Gesù di Roma.
Il gene responsabile dell’atrofia muscolare spinale (SMA) ha effetti sul metabolismo degli aminoacidi del cervello e del fegato fin dai primi giorni dalla nascita. Incide anche sull’espressione degli enzimi che permettono la sintesi dei neurotrasmettitori, cioè delle molecole segnale usate nella comunicazione tra le cellule nervose.
«Un risultato importante – commenta l’ideatore del progetto Alessandro Usiello, Direttore del Laboratorio di Neuroscienze Traslazionali del CEINGE e professore di Biochimica Clinica dell’Università Vanvitelli – che da un lato fa pensare alla possibilità di stabilire nuovi biomarcatori per predire l’esordio della malattia, dall’altro suggerisce l’importanza della nutrizione per compensare i deficit metabolici causati dalla riduzione della proteina SMN».
«Abbiamo studiato – spiega ancora Usiello – il profilo metabolomico, epatico e cerebrale, di modelli animali SMA nelle diverse fasi della malattia. I risultati hanno svelato che nella SMA è presente una notevole alterazione metabolica di numerosi amminoacidi, accompagnati da una severa riduzione dei livelli del neurotrasmettitore noradrenalina, implicato anche nella regolazione dell’eccitazione delle cellule nervose, del loro metabolismo energetico e delle risposte infiammatorie».
«I risultati ottenuti nei modelli murini SMA – chiarisce Francesco Errico, ricercatore del CEINGE e professore di Biochimica generale presso il Dipartimento di Agraria dell’Università Federico II – sono coerenti con quanto abbiamo riscontrato nel liquido cerebrospinale dei pazienti pediatrici SMA 1 dell’ospedale Bambino Gesù, nei quali si evidenziano alterazioni nel metabolismo di diversi amminoacidi, nonché un aumento dei livelli di noradrenalina dopo il trattamento col Nusinersen, noto aumentare i livelli di SMN».
Secondo i neurologi dell’Ospedale pediatrico romano Bambino Gesù, Enrico Bertini ed Adele D’Amico, lo studio è molto promettente: «Oltre ad aver finalmente iniziato a fare luce sulla complessa rete di alterazioni metaboliche e neurometaboliche esistenti nella SMA, i risultati della ricerca suggeriscono possibili approcci terapeutici e nutrizionali combinati e personalizzati, potenziando le terapie oggi disponibili».
Le ricerche, svolte in collaborazione con i gruppi di ricerca di Pino Pignataro, professore di Farmacologia dell’Università Federico II, di Manolo Carta, professore di Fisiologia dell’Università di Cagliari, e di Anna Maria D’Ursi, professore di Chimica Farmaceutica dell’Università di Salerno, sono state in parte finanziate dai Fondi PNRR project MNESYS–A Multiscale integrated approach to the study of the nervous system in health and disease.
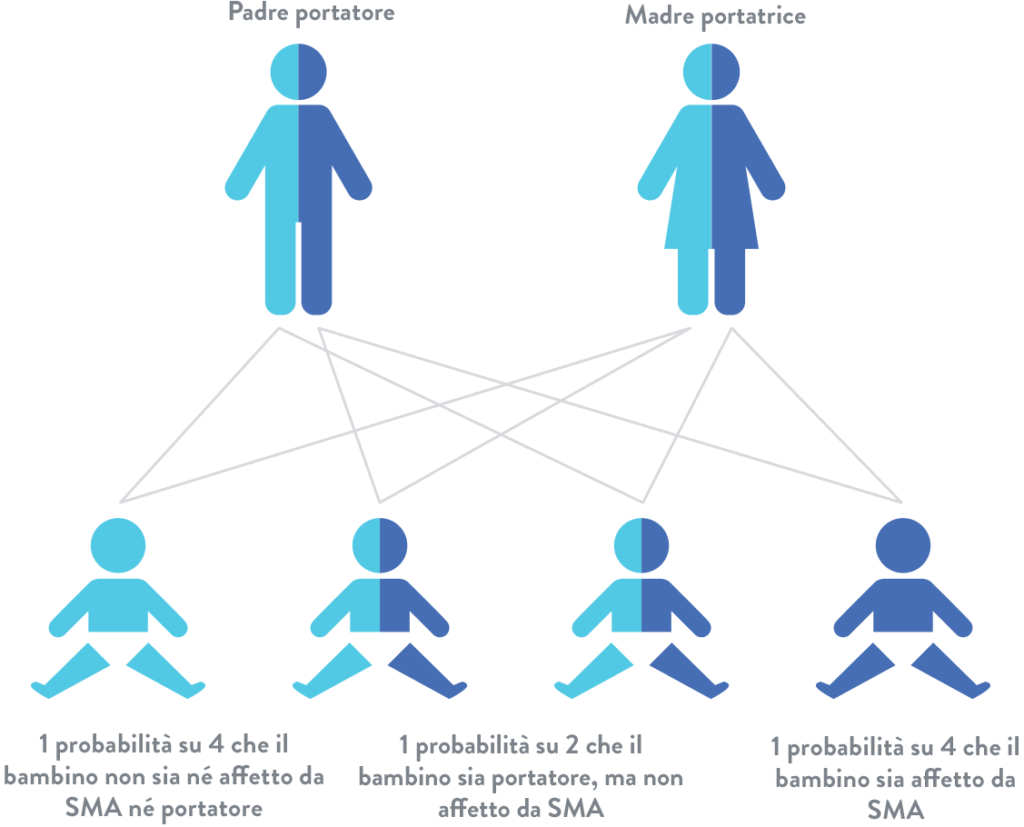
L’atrofia muscolare spinale (SMA) è una patologia neuromuscolare rara molto spesso diagnosticata durante l’infanzia, contraddistinta dalla precoce morte dei motoneuroni, ovvero le cellule nervose che trasportano i segnali dal sistema nervoso centrale ai muscoli, controllandone la struttura, la forza e il movimento. Per questo motivo, la malattia determina atrofia muscolare progressiva e debolezza che colpisce, in modo preponderante, gli arti inferiori e i muscoli respiratori. La SMA ha un’incidenza di circa 1 paziente su 11mila nati. Nel 95% dei casi, la patologia è causata da specifiche mutazioni nel gene SMN1, che codifica per la proteina SMN (Survival Motor Neuron), essenziale per la sopravvivenza e il normale funzionamento dei motoneuroni. Attualmente, sono state approvate tre terapie specifiche per questa malattia: l’oligonucleotide antisenso Nusinersen, che deve essere somministrato per via intratecale, e il farmaco orale Risdiplam, progettati per agire sul gene SMN2 e permettere la produzione di una proteina SMN funzionale, e la terapia genica onasemnogene abeparvovec, concepita per fornire all’organismo una versione sana del gene SMN1. Nonostante le recenti terapie abbiano notevolmente migliorato la qualità della vita dei pazienti SMA, soprattutto quando trattati prima dell’esordio dei sintomi, appare chiaro che nessuna di queste rappresenta una cura definitiva per la malattia. Infatti, la correzione incompleta dei sintomi della malattia, unita alla variabilità della risposta clinica al trattamento rappresentano ancora un limite che sembra per ora invalicabile.
SMA, identificate le alterazioni neurometaboliche che interferiscono con la comunicazione tra le cellule nervose
I risultati di una ricerca italiana aprono la strada a nuovi approcci terapeutici per potenziare i farmaci oggi disponibili
Un network di ricercatori e clinici nel campo delle neuroscienze ha identificato gravi alterazioni biochimiche epatiche e neurometaboliche nella Atrofia Muscolare Spinale (SMA), una malattia genetica rara, che nella forma più grave provoca paralisi fino alla morte prematura dei bambini. I risultati dello studio, durato 4 anni e condotto su 33 pazienti pediatrici ed in modelli animali, sono stati pubblicati sulla rivista scientifica Communication Biology (Nature Group). La ricerca è stata sviluppata grazie ad una stretta collaborazione tra il CEINGE Biotecnologie Avanzate Franco Salvatore di Napoli, l’Università della Campania Luigi Vanvitelli, l’Università di Napoli Federico II, l’Università di Salerno, l’Università di Cagliari e l’Ospedale Bambino Gesù di Roma.
Il gene responsabile dell’atrofia muscolare spinale (SMA) ha effetti sul metabolismo degli aminoacidi del cervello e del fegato fin dai primi giorni dalla nascita. Incide anche sull’espressione degli enzimi che permettono la sintesi dei neurotrasmettitori, cioè delle molecole segnale usate nella comunicazione tra le cellule nervose.
«Un risultato importante – commenta l’ideatore del progetto Alessandro Usiello, Direttore del Laboratorio di Neuroscienze Traslazionali del CEINGE e professore di Biochimica Clinica dell’Università Vanvitelli – che da un lato fa pensare alla possibilità di stabilire nuovi biomarcatori per predire l’esordio della malattia, dall’altro suggerisce l’importanza della nutrizione per compensare i deficit metabolici causati dalla riduzione della proteina SMN».
«Abbiamo studiato – spiega ancora Usiello – il profilo metabolomico, epatico e cerebrale, di modelli animali SMA nelle diverse fasi della malattia. I risultati hanno svelato che nella SMA è presente una notevole alterazione metabolica di numerosi amminoacidi, accompagnati da una severa riduzione dei livelli del neurotrasmettitore noradrenalina, implicato anche nella regolazione dell’eccitazione delle cellule nervose, del loro metabolismo energetico e delle risposte infiammatorie».
«I risultati ottenuti nei modelli murini SMA – chiarisce Francesco Errico, ricercatore del CEINGE e professore di Biochimica generale presso il Dipartimento di Agraria dell’Università Federico II – sono coerenti con quanto abbiamo riscontrato nel liquido cerebrospinale dei pazienti pediatrici SMA 1 dell’ospedale Bambino Gesù, nei quali si evidenziano alterazioni nel metabolismo di diversi amminoacidi, nonché un aumento dei livelli di noradrenalina dopo il trattamento col Nusinersen, noto aumentare i livelli di SMN».
Secondo i neurologi dell’Ospedale pediatrico romano Bambino Gesù, Enrico Bertini ed Adele D’Amico, lo studio è molto promettente: «Oltre ad aver finalmente iniziato a fare luce sulla complessa rete di alterazioni metaboliche e neurometaboliche esistenti nella SMA, i risultati della ricerca suggeriscono possibili approcci terapeutici e nutrizionali combinati e personalizzati, potenziando le terapie oggi disponibili».
Le ricerche, svolte in collaborazione con i gruppi di ricerca di Pino Pignataro, professore di Farmacologia dell’Università Federico II, di Manolo Carta, professore di Fisiologia dell’Università di Cagliari, e di Anna Maria D’Ursi, professore di Chimica Farmaceutica dell’Università di Salerno, sono state in parte finanziate dai Fondi PNRR project MNESYS–A Multiscale integrated approach to the study of the nervous system in health and disease.
L’atrofia muscolare spinale (SMA) è una patologia neuromuscolare rara molto spesso diagnosticata durante l’infanzia, contraddistinta dalla precoce morte dei motoneuroni, ovvero le cellule nervose che trasportano i segnali dal sistema nervoso centrale ai muscoli, controllandone la struttura, la forza e il movimento. Per questo motivo, la malattia determina atrofia muscolare progressiva e debolezza che colpisce, in modo preponderante, gli arti inferiori e i muscoli respiratori. La SMA ha un’incidenza di circa 1 paziente su 11mila nati. Nel 95% dei casi, la patologia è causata da specifiche mutazioni nel gene SMN1, che codifica per la proteina SMN (Survival Motor Neuron), essenziale per la sopravvivenza e il normale funzionamento dei motoneuroni. Attualmente, sono state approvate tre terapie specifiche per questa malattia: l’oligonucleotide antisenso Nusinersen, che deve essere somministrato per via intratecale, e il farmaco orale Risdiplam, progettati per agire sul gene SMN2 e permettere la produzione di una proteina SMN funzionale, e la terapia genica onasemnogene abeparvovec, concepita per fornire all’organismo una versione sana del gene SMN1. Nonostante le recenti terapie abbiano notevolmente migliorato la qualità della vita dei pazienti SMA, soprattutto quando trattati prima dell’esordio dei sintomi, appare chiaro che nessuna di queste rappresenta una cura definitiva per la malattia. Infatti, la correzione incompleta dei sintomi della malattia, unita alla variabilità della risposta clinica al trattamento rappresentano ancora un limite che sembra per ora invalicabile.